Background
Methylation sequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA to determine its pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
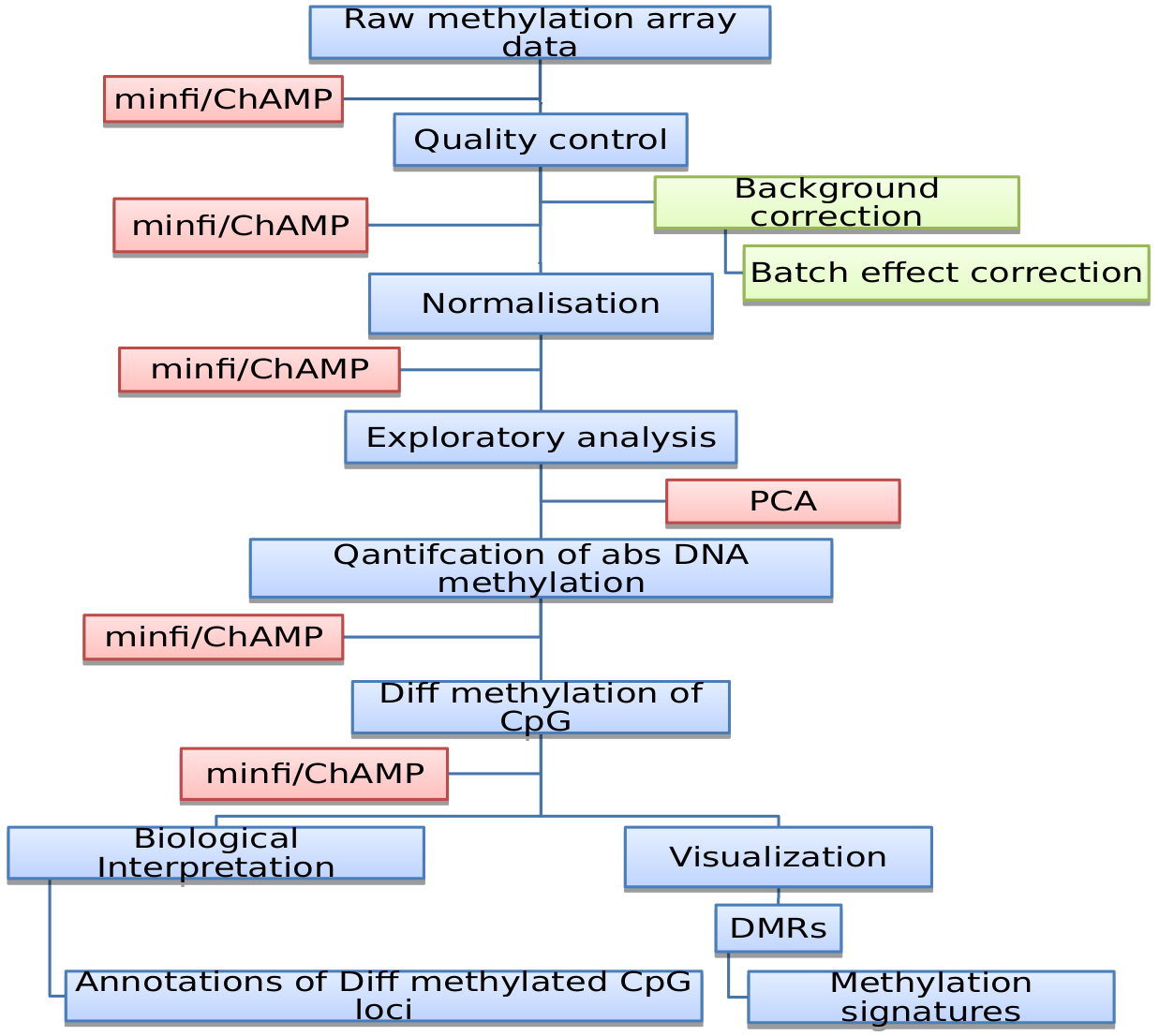
Genome-wide DNA methylation is mapped with one of the three most commonly used assays, resulting in methylation-specific DNA sequencing or microarray data (CpG methylation array).
Figure: CpG methylation array analysis workflow