Background
DNA sequencing is the process of determining the precise order of nucleotides within a DNA molecule. It includes any method or technology that is used to determine the order of the four bases Adenine, Guanine, Cytosine and Thymine in a strand of DNA. The advent of rapid DNA sequencing methods enabled variants discovery (SNP, Insertion, deletion, CNV etc.), which has greatly accelerated biological and medical research.
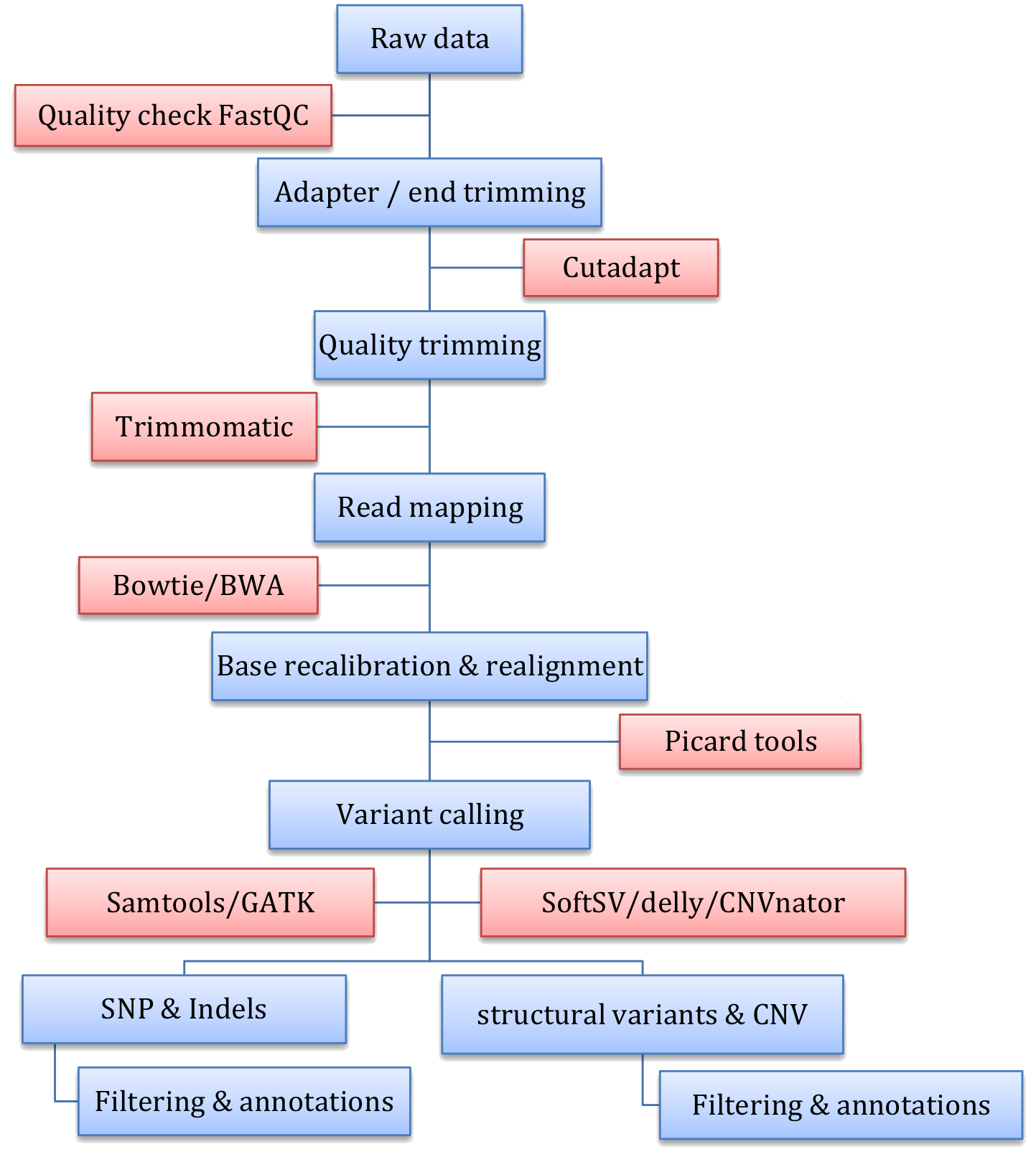
Figure: DNA seq analysis workflow