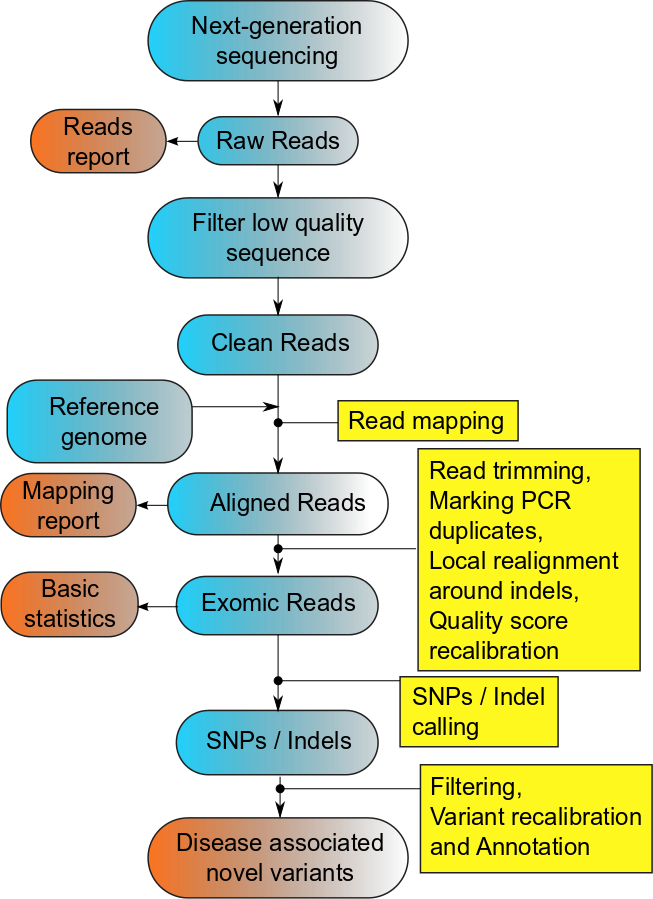
For analyzing sequence data, alignment to a reference genome is necessary. For humans there are
several options of retrieving the reference sequence: the most important being UCSC and Ensembl.
For aligning the sequences to the human genome are used different read aligners.
BWA, Bowtie, etc. are the ultrafast and memory-efficient tools for aligning sequencing
reads to long reference sequences.
It needs very few memory, works well with Illumina data and can be run using several threads.
For using the read aligners, the reference genome needs to be indexed and transformed
using an indexing program. That may take a while. The result is a SAM file which is needed for later analysis.
The SAM file is the starting point for obtaining a much more powerful file type: the binary
Alignment/Map format (short BAM). It compresses the SAM file and can be indexed, which means
that only portions of the file can be accessed without the need to load the whole file.
A usual exome BAM file takes about 4-10 Gb disk space while a SAM file needs 20-30 Gb.
For converting the SAM file to BAM we use picard. Picard offers many options for manipulating
or viewing SAM and BAM files.