Background
RNA-Seq (RNA sequencing), also called whole transcriptome shotgun sequencing (WTSS), uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA (gene expression) and isoforms variants in a biological sample at a given moment in time.
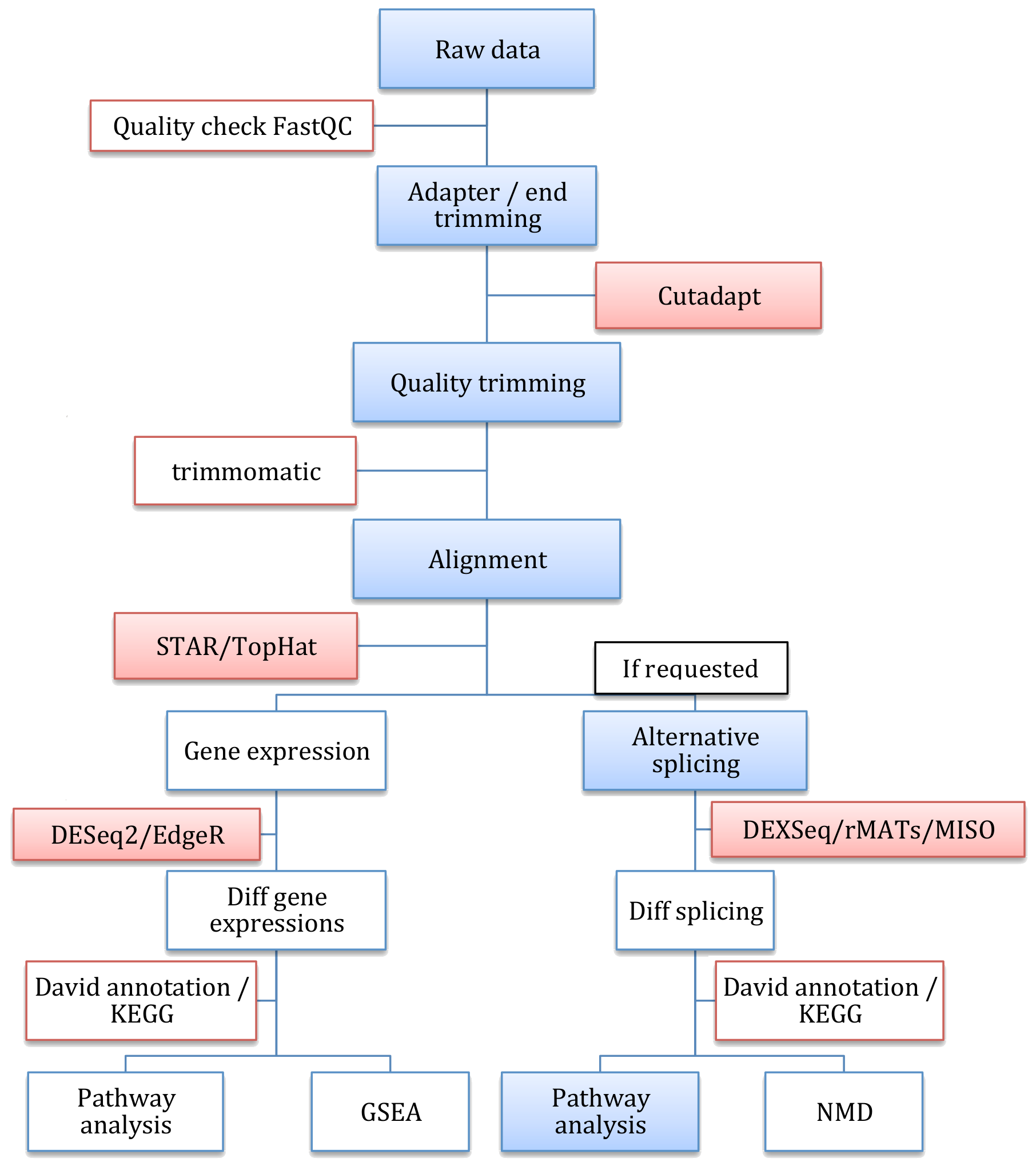
Figure: RNA-Seq workflow